AlphaFold 기반 신약개발 연구 전략


SBDD(Structure-Based Drug Design)
이전 글에서 CADD (Computer-Aided Drug Design)는 크게 SBDD(Structure-Based Drug Design)와 LBDD(Ligand-Based Drug Design)로 나눌 수 있다고 말씀드렸습니다.

SBDD : 장점과 단점
SBDD는 신약개발의 표적이 되는 단백질의 3차원 구조 정보를 기반으로 진행됩니다. 대량의 화합물 라이브러리에서 표적 단백질에 안정적으로 결합하는 분자 구조(유효 물질)를 찾는 virtual screening이나 확보한 유효 물질이 더 잘 결합할 수 있도록 분자 구조를 최적화하는 hit/lead optimization 등이 대표적인 SBDD 과정입니다. 약물의 활성이 표적 단백질과의 상호작용의 결과라는 점에서 3차원 공간에서의 binding pocket의 형태나 단백질-약물 상호작용을 반영할 수 있는 SBDD는 매우 강력한 도구라 할 수 있습니다.
이러한 3차원 단백질 구조의 사용은 동시에 SBDD의 가장 큰 약점이 되기도 합니다. SBDD의 핵심은 단백질과 화합물의 3차원 결합 구조에 대한 결합 에너지(활성) 예측이며, 당연하게도 올바른 단백질-화합물 결합 구조를 입력 정보로 사용해야 정확한 예측 결과를 얻을 수 있습니다. 아쉽게도 이러한 결합 구조가 실험으로 결정된 경우는 거의 없기 때문에 docking을 통해 이를 예측하는 과정을 거치게 됩니다. 아래 그림은 docking 과정에서 고정된 단백질 구조에 화합물의 구조(conformation)를 바꿔가며 단백질-화합물 구조를 생성하는 과정을 보여주고 있습니다.

SBDD : Docking 과정 및 Scoring Function
Docking 과정에서는 단백질 구조를 고정한 상태에서 화합물의 conformation을 다양하게 바꿔가며 결합 구조를 탐색합니다. 이때 화합물과 단백질의 상호작용을 평가하여 점수를 메기는 scoring function이 사용되며 보통 10개 전후의 상위 결합 구조를 제공합니다. Docking 과정에서 단백질 구조를 고정하는 것은 계산 효율 측면에서는 큰 도움이 되지만 정확성을 제한하는 원인이 되기도 합니다. 단백질의 구조는 약물의 결합이나 다른 단백질과의 상호작용에 의해 달라질 수 있기 때문입니다. 이는 고정된 단백질 구조를 사용하는 docking 과정에서 단백질 구조를 신중히 선택해야 한다는 것을 의미합니다.
표적 단백질의 실험 구조가 없는 경우
만약 표적 단백질의 실험 구조가 없는 경우에는 어떨까요? 고전적으로는 homology modeling 등의 방법을 사용해서 예측한 단백질 구조를 사용합니다. 이 경우 일정 수준의 sequence identity(보통 30% 이상)를 가진 단백질의 3차원 구조가 필요하다는 한계와 MD simulation등의 방법으로 추가적인 최적화를 진행해야 한다는 등의 어려움이 여전히 남아 있습니다.

AlphaFold의 등장이 시사하는 점
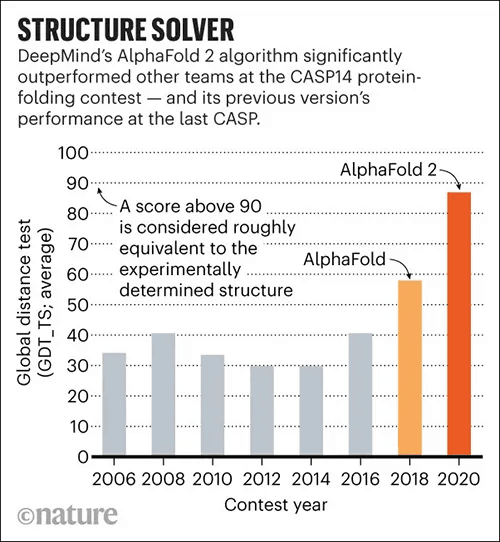
단백질 구조 예측은 오랜 시간 많은 관심을 받아온 분야이며 AlphaFold 등장 이전까지는 절대 강자가 없는 춘추전국시대였습니다. AlphaFold는 2018년 갑작스럽게 등장하여 기존 방법들을 압도하였고 2020년 이 분야를 평정하였습니다. 아래 그림을 보면 AlphaFold, 특히 AlphaFold2의 경우 절대적인 성능을 가지고 있으며 GDT가 90에 가까워 예측 결과가 실험 구조에 근접하는 정확성을 확인 할 수 있습니다.
AlphaFold 단백질 구조 예측의 한계점
그렇다면 이제 AlphaFold가 예측한 단백질 구조를 사용해서 모든 문제가 해결할 수 있는 걸까요? AlphaFold는 단백질만을 고려해서 구조를 예측하기 때문에 다음 3가지 한계가 있습니다.
- 약물이 결합하는 위치인 binding site의 위치 정보가 없습니다. AlphaFold 구조는 보통 어떤 상황에서 사용하게 될까요? 주로 데이터 확보가 어려운 first-in-class drug 연구에서 사용 할 것으로 생각됩니다. Best-in-class 등의 경우에는 표적 단백질 구조나 활성 데이터가 축적되어 있어 굳이 AlphaFold 구조를 사용할 이유가 없으니까요. 다른 정보 없이 AlphaFold 구조만으로 binding site를 정의하거나 핵심 상호작용 등을 찾는 것은 쉽지 않은 과정입니다.
- 단백질은 임 없이 움직이는 유동적인 구조라는 점을 다시 한번 생각해 봅시다. 화합물이 단백질에 결합할 경우 단백질 구조에 크고 작은 영향을 미치게 됩니다. AlphaFold는 단백질만 있는 구조 (Apo form)이기 때문에 화합물의 결합이 단백질 구조에 영향을 줄 경우 적합하지 않은 구조입니다.
- 금속이나 ATP와 같은 cofactor가 있는 경우에는 어떻게 해야 할까요? Binding site와 거리가 있다면 다행이지만 가까울 경우 약물과 상호작용을 할 가능성이 있기 때문에 적절한 위치에 cofactor를 배치해야 합니다.
AlphaFold를 활용한 신약 개발 정보 확보
자 그럼 이제부터는 AlphaFold 예측 구조 정보만 가지고 있는 상황에서 어떤 방식으로 신약개발 연구를 위한 정보를 확보하기 위한 전략을 알아보도록 하겠습니다. 전체적인 과정은 아래 그림과 같습니다.
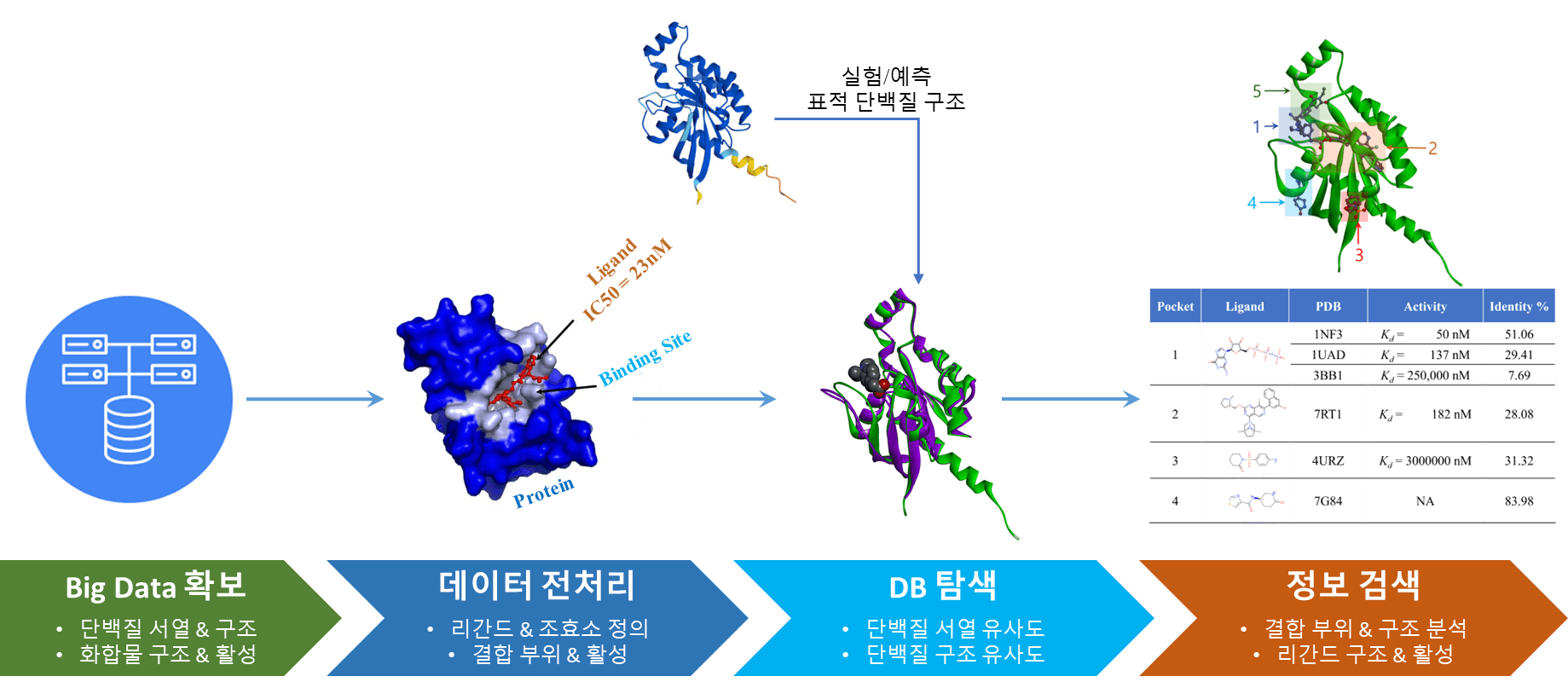
이 과정은 대량의 정보 처리 작업이 포함되어 있어 수작업으로는 진행하기 쉽지 않습니다. 신약개발 연구자가 쉽게 사용할 수 있도록 자동화하여 하이퍼랩에 추가 할 예정이니 많은 기대 바랍니다! 하이퍼랩에 추가되기 전에 사용이 필요하시다면 연락 주시기 바랍니다.
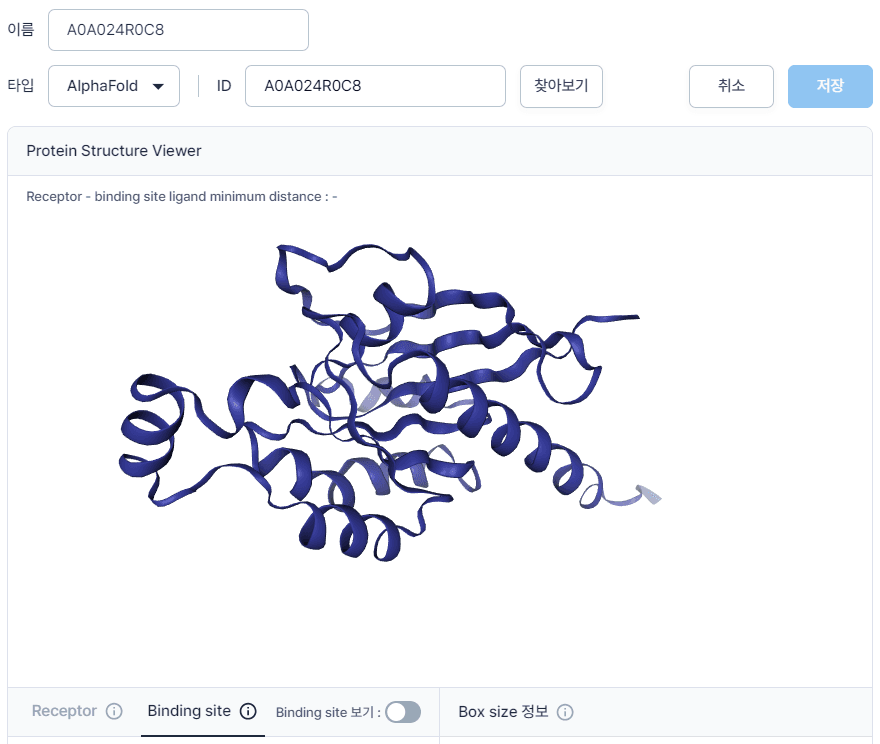
1. 가장 먼저 표적 단백질의 AlphaFold 구조를 확보하겠습니다. 예제는 RCSB에 구조 정보가 없는 Epididymis secretory sperm binding protein (UniProt ID: A0A024R0C8)을 표적 단백질로 사용하겠습니다. AlphaFold 웹페이지에서 “Epididymis secretory sperm binding protein”검색하면 매우 많은 검색 결과를 확인 할 수 있습니다. 원하는 단백질을 정확하게 검색하기 위해서는 UniProt ID를 사용할 것을 추천 합니다. UniProt ID는 UniProt 홈페이지에서 확인 할 수 있습니다. UniProt ID A0A024R0C8 검색하여 AlphaFold 구조를 다운로드합니다.
하이퍼랩에서는 “Protein Structure” 탭에서 “타입”을 “AlphaFold”로 선택하고 UniProt code를 입력하시면 손쉽게 단백질 구조를 얻으실 수 있습니다.
참고로 UniProt 정보에 따르면 이 단백질은 GTPase activity가 있고 small GTPase mediated signal transduction에 관여한다고 합니다.

2. 이제 RCSB에서 AlphaFold 예측 구조와 유사한 단백질 구조를 찾아봅시다! 여기서부터는 대량의 데이터를 다루기 때문에 수작업으로 진행하기가 거의 불가능합니다. 간단하게 설명하고 결과에 집중하도록 하겠습니다. 단백질 구조의 유사도를 확인하기 위해서는 우선 두 단백질을 정렬(sequence & structure alignment)한 후 유사한 정도를 평가합니다. 유사도는 매칭되는 두 단백질의 아미노산 (보통 CA 원자) 사이의 RMSD를 계산하거나 TM-score, p-value 등을 사용할 수 있습니다.

이러한 과정을 통해 A0A024R0C8와 유사한 PDB 구조를 약 1200개 확보 하였습니다. 검색된 단백질들은 KRAS, HRAS, Rap1, RHO 등 표적 단백질과 기능적으로, 그리고 구조적으로 유사한 단백질로 확인됩니다.
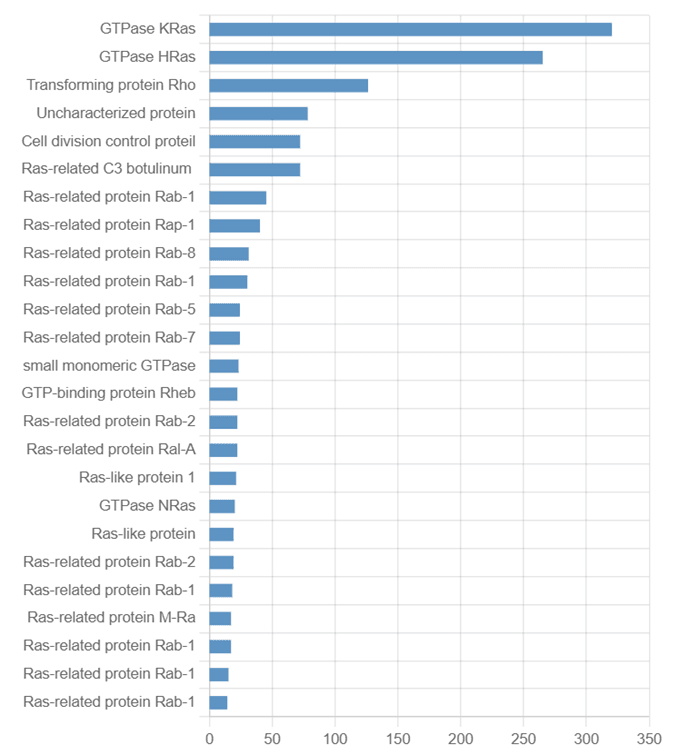
구조 유사성이 높은 단백질 구조들을 확보했다면 이제 이 구조들에 존재하는 inhibitor 등의 ligand를 정의할 차례입니다. Ligand는 실험 논문 등을 통해 하나하나 확인하는 것도 가능하겠지만 현실적으로 무리입니다. RCSB에는 20만 개가 넘는 PDB 구조가 저장되어 있으니까요. 다른 방법은 PDBBind, BindingDB 등의 DB를 사용하는 방법입니다. RCSB PDB 구조에 포함된 ligand들의 활성 데이터가 정리되어 있어 상당 부분의 ligand를 정의 할 수 있습니다. 히츠에서는 최대한의 실험 정보를 수집하여 리간드와 cofactor에 대한 DB를 구축하고 있으며, 이를 활용하여 하이퍼랩에서 자동으로 binding site를 정하는 기능을 제공하고 있습니다.
데이터 기반의 정보 확보 및 분석
이제 유사도가 높은 단백질 구조와 이 구조에 존재하는 활성 분자들의 정보를 확보 했습니다. 이제 남은 과정은 AlphaFold 구조에 이 정보를 대입하는 것입니다. 이러한 과정을 통해 binding site일 가능성이 높은 위치를 찾을 수 있습니다. 아래 그림은 DB에서 검색된 유사 구조 단백질과 그들의 binding site 및 리간드 활성 정보를 정리한 것입니다.
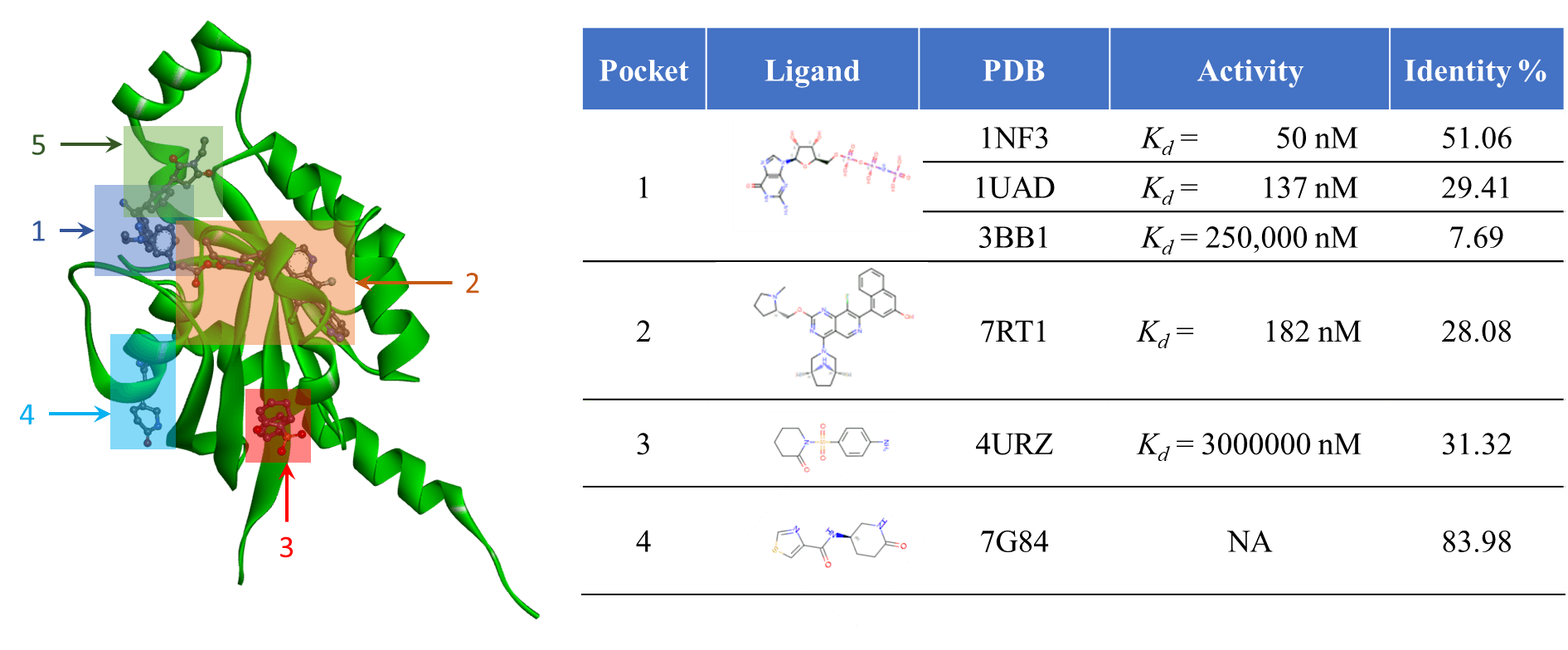
위 그림에서는 5개의 binding site일 가능성이 높은 위치와 각 site에 대한 근거 데이터를 보여주고 있습니다. 물론 이 정보들을 우리가 관심을 가진 단백질에 100% 적용 할 수 있는 것은 아닙니다. 주어진 데이터를 분석하여 옥석을 가려낼 필요가 있습니다. 위 그림에서 1번 site에 결합하는 물질의 경우 1NF3에서는 kd = 50nM로 매우 높은 활성을 보이지만, 3BBI에서는 250,000nM로 활성이 없는 것을 확인 할 수 있습니다. 같은 분자인데 왜 이렇게 활성에 큰 차이가 있는 걸까요? 서로 다른 두 단백질의 binding site를 구성하는 잔기(residue)들이 유사할 수록 한 단백질의 ligand가 다른 단백질에서도 유사한 활성을 가질 것이라 직관적으로 알 수 있을 것 입니다. A0A024R0C8에 대한 1NF3와 3BB1의 sequence identity는 각각 51.06%와 7.69%로 매우 다르라는 점에서 활성 차이를 간접적으로 이해 할 수 있습니다. 아래 그림은 결합 위치의 잔기들을 두 단백질에서 같을 경우 푸른색, 다를 경우 붉은 색으로 보여주고 있습니다.

1NF3의 경우 A0A024R0C8와 pocket을 구성하는 잔기가 거의 같지만 3BB1의 경우 이와는 반대로 매우 다른 것을 알 수 있습니다. 이 경우 당연히 pocket이 비슷한 1FN3 정보를 사용하는 것이 맞을 것 같습니다.
데이터 기반의 정보 확보 및 분석은 DB에 정리된 정보나 사용 목적에 따라 달라질 수 있습니다. 예를 들어 결합 부위를 구성하는 잔기나 골격의 구조적 유동성 분석을 통해 AlphaFold 구조를 최적화하는 등의 전략을 사용할 수 있습니다.
마무리
SBDD와 단백질 구조 예측 기술인 AlphaFold는 현대 신약 개발 분야에서 중요한 도구로 자리 잡고 있습니다. 그러나 이러한 기술들은 여전히 한계를 갖고 있으며, 완벽한 해결책이라기보다는 연구 및 개발 과정에서 도움을 주는 도구로서 활용되고 있습니다. 앞으로의 연구와 개발 과정에서는 SBDD와 단백질 구조 예측 기술을 비롯한 다양한 도구들을 유기적으로 결합하여 새로운 치료제의 발견과 개발에 기여할 수 있기를 기대합니다.